9月11日,CFDA发布《已上市中药生产工艺变更研究技术指导原则》(简称“正式稿”,下同)的通告。在中药正式稿发布前, CFDA于8月29日发布了《已上市化学药品生产工艺变更研究技术指导原则》。9月7日,CDE开始网上征求《生物制品上市后变更研究技术指导原则》意见。
如此,三类药品的工艺变更研究技术指导原则有望在2017年全部发布完毕。而随着工艺变更指导原则全部发布,CFDA飞检就有了理论依据。工艺核对的飞检,对于企业来说关乎生存,因此各生产企业应尽快开展工艺变更研究。
工艺变更飞检已经开始
2016年8月9日,CFDA公开征求《关于开展药品生产工艺核对工作的公告(征求意见稿)》的意见,原定于2016年9月10日修改意见征集完毕,药品生产企业于2016年10月1日前完成自查并将自查情况报所在地省级食品药品监管部门。省级食品药品监管部门应对企业自查情况进行汇总,填写自查情况汇总表并于2016年11月1日前上报食品药品监管总局。
按征求意见稿,药品生产企业应于2017年6月30日前完成在产品种生产工艺的研究验证、实际生产工艺与批准生产工艺不一致的提交补充申请等相关工作,其他暂不生产品种应于2017年12月31日前完成上述工作;未按时完成的,2017年6月30日停止生产。
此外,2016年11月1日起,CFDA会组织专家对药品生产企业针对生产工艺开展飞检。如表1所示,2016年12月CFDA在飞检时就已经开始检查工艺了。
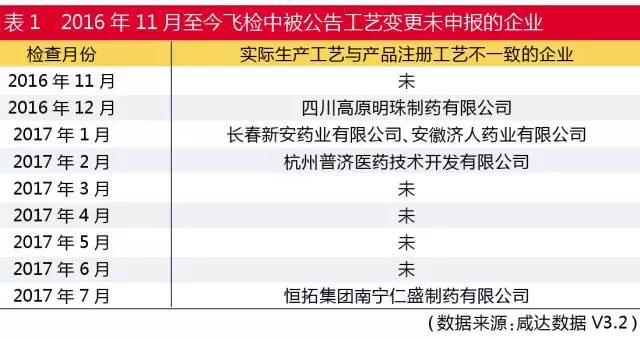
以2016年12月9-11日接受飞检的四川高原明珠制药有限公司为例,CFDA公告该企业复方板蓝根颗粒生产批量由5万袋变更为48万袋,提取设备变更前使用1个提取罐,目前使用4个提取罐生产,未进行变更控制,且未对变更的批量进行风险评估和工艺验证。由此可见,工艺变更已经是CFDA的飞检项目内容之一了。
中药“正式稿”九个不同细节
2017年3月,CDE网站发布关于《已上市中药生产工艺变更研究技术指导原则》征求意见(简称“征求意见稿”,下同)的通知。咸达数据V3.2对“征求意见稿”与刚刚发布的“正式稿”进行比较,发现主要有以下9个细节变更。
1.申请人对已上市中药拟变更生产工艺开展研究,“征求意见稿”中的“并向药品监督管理部门提出补充申请”字眼被删去。
2.认可工艺变更往更好的质量方向提升。“正式稿”要求“无论何种类别的变更,都不应对药品的安全性、有效性发生负面影响”,“征求意见稿”原文是“不允许安全性、有效性发生明显改变”。
3.中药生产工艺变更研究的质量方法举例,除了“征求意见稿”提到的指纹图谱(特征图谱)和溶出度检查以外,“正式稿”增加生物活性检测。中药生产工艺变更研究及申报资料要求的Ⅰ类变更的“质量对比研究”中,“正式稿”也增加了生物活性检测。
4.研究验证样品从“中试以上规模”改为“能够代表生产实际情况的”,中药生产工艺变更研究及申报资料要求的Ⅰ类变更中对“中试生产数据”的要求也从“提供3批稳定的中试研究数据”改为“提供3批能够代表生产实际情况的中试研究数据”。
这基本与目前注册法规大趋势一致,例如新化学药分类注册资料要求生产工艺以目前生产的最大批量。
5.药材前处理变更分类的Ⅰ类变更删除了“不含热敏性、挥发性成分的药材或原粉增加瞬间高温灭菌处理步骤”。
6.提取纯化的变更分类的Ⅰ类变更中的“药液静置、过滤”改为离心;药液中的“固形物含量不变”改为“药液静置”,过滤改为离心(或离心改为药液静置、过滤);药液中的“固形物及指标成份含量不变”,“正式稿”对指标含量的变化有要求。
成型工艺变更分类的Ⅰ类变更,对于增加药液普通过滤工序,或者变更药液普通过滤的滤材材质、孔径及过滤次数等工序,“正式稿”也新增要求“相关检测及固形物、指标成份含量等不变”。
7.成型工艺变更分类的Ⅲ类变更,“正式稿”将“对药物理化性质等有明显影响的成型工艺方法的改变”改为“对药物吸收利用等有明显影响的成型工艺方法的改变”,更关注药物动力学相关内容。
8.成型工艺变更分类的Ⅲ类变更中,“正式稿”删去“缓释/控释制剂中缓释材料种类或用量变更”。
中药生产工艺变更研究及申报资料要求的Ⅲ类变更方面,删去“缓释/控释制剂变更辅料属于Ⅲ类变更者,应提供药代动力学研究资料”和“外用制剂变更辅料属于Ⅲ类变更者,需要提供制剂非临床安全性研究资料”两项内容。
9.中药生产工艺变更研究及申报资料要求的Ⅱ类变更方面,临床试验或生物等效性研究比较资料正式稿删去“其中临床试验研究进行病例数不少于100对的临床试验,用于多个病证的,每一个主要病证病例数不少于60对”的临床试验限制。
哪些企业进行工艺变更备案?
咸达数据V3.2分析药品注册补充申请备案情况公示数据发现,截至2017年9月13日,2016年8月以后与工艺变更相关的补充申请共74条。74条基本都是不改变产品质量的生产工艺变更提出备案申请。
备案机构方面,73条属于重庆市食品药品监督管理局,1条属于陕西省食品药品监督管理局。重庆的工艺变更补充申请备案数之所以排名第一,是因为2016年3月重庆发布了《重庆市食品药品监督管理局关于加强药品生产工艺及处方清理工作的补充通知》(渝食药监药生产[2016]14号)。药品生产企业提出无质量差别的变更申请后,经重庆市药品技术审评认证中心对申报资料审定,变更内容不影响产品内在质量,才能予以备案。
生产企业方面肯定也是重庆的企业包揽前三。福安药业(含庆余堂制药)受理号数最多,有37个;其次是天圣制药,32个,重庆赛维药业有限公司以2个排第三。
药品分类方面,化学药品72条工艺变更,生物制品和中药各1条。化学药品变更的产品以抗生素的注射液和原料药为主。生物制品变更工艺的是酪酸梭菌活菌胶囊,中药则是薏辛除湿止痛胶囊。
小结<<<
从重庆的数据可推测,各企业对工艺变更的申报主要为“不改变产品质量的生产工艺变的备案补充申请”,预计一些工艺技术较成熟的注射剂会较容易获批备案。
2016年临床自查核查以来,大批量的申报被撤回,个别企业对于工艺核对的申报有点草木皆兵,生怕被秋后算账。毕竟临床自查核查对企业来说,属于新产品开发受挫;一致性评价则可视为产品在医疗机构采购渠道受限;工艺核对对于企业来说可谓生存之战,一旦飞检不能通过,原有的产品可能被停产做工艺变更研究,GMP资质还有被取消的风险,企业岂能不重视?
随着工艺变更指导原则全部发布,为CFDA飞检提供了理论依据。各生产企业要尽快开展工艺变更的研究了。




 相关新闻
相关新闻














