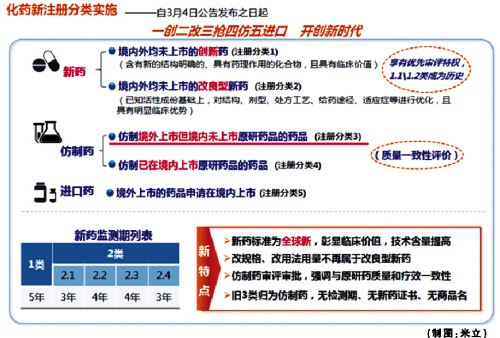
新3类药回归本位,一致性评价定未来
在CFDA出台的《化学药品注册分类改革工作方案》中,新的药品注册管理办法给新药做了重新定义。新药的概念从“中国新”上升到“全球新”,而3类药自此被纳入仿制药范畴。值得注意的是,大量申请老3类药的企业将面临巨大挑战,已进入临床阶段的老3类药物和处于注册申请受理阶段的老3类药物何去何从?企业该如何抉择?
业内人士表示,期待尽快出台关于新3类药的临床试验要求,以及新老3类药物在政策执行上如何过渡等方面的详细指导原则。
回归仿制药本位
新3类药物申报降温
尽管老3类药本质上属于仿制药,但在“新药”头衔的庇护下享有3-4年新药监测期,即未到期前CDE不再受理相关进口和国产的注册申请,这也使得很多企业在合成工艺、处方工艺等研究不充分的条件下盲目抢夺首仿,很长一段时间内老3类药申报竞争非常激烈。
在新的注册分类改革下,以首仿药、抢仿药为策略的国内企业将不得不考虑新的出路,同时新3类药物申报热将大幅降温。不过,也有分析认为抢仿以市场为导向的临床优势品种依旧会迎来激烈竞争。
某药业副总经理告诉记者,老3类药物未来不仅面临原研药进口的压力,还要面临有所提升的仿制药申报门槛。“企业需要对自身实力进行评估,比如生产工艺按照现在颁布的分类要求和指导原则对比原研药能否达到等效,如果达不到标准,可能就要考虑重新做甚至是放弃。”
“化学药品新注册分类申报资料对生产工艺、过程控制、物料控制、关键步骤和中间体的控制都有明确的要求,企业如果要开发新3类药物,除了在技术上要有强大的支撑外,还需要投入更多的人力、物力和财力,如参比制剂的购买,质量标准的购买以及短期内较多的人员沟通。”某生物医药产业技术研究院有限公司总经理认为,新政策总体要求更严格,很多企业不会仅仅看中产品本身市场而进行仿制,而是更多地考虑企业本身的战略规划、投资、研发实力等等。
有专家向记者坦言,“以往很多研发公司出售的都是3类药物临床批件,前年还能查到有价值几百万元的临床批件转让,随着一致性评价等政策门槛的提高,药品注册费用上涨,现在这类临床批件的转让价格已经大幅度缩水了。”
出现这种现象的根本原因在于,新3类药定义中强调了“应与原研药品的质量和疗效一致”的概念。企业无论是转让还是再开发,都无法逃避与原研药进行一致性评价这道门槛。
老3类药何去何从?
随着《仿制药质量和疗效一致性评价工作程序(征求意见稿)》和《化学药品仿制药口服固体制剂一致性评价申报资料要求(征求意见稿)》等文件的下发,在一致性评价大势所趋的背景下,未来三年补充申请的数量会大幅上升,而数量上升又会造成一定程度上的积压,同时投入的成本和未来市场同样会令企业权衡,企业会做出何种决择呢?
记者在采访中了解到,对于已经注册获得临床批件的老3类药物,企业的当务之急是评估产品的临床价值和市场价值,进而判断是否有继续下去的必要。有专家根据已公布的政策分析认为,“应该是BE和100对随机临床对照试验都要做的,按照现在临床数据的核查情况,企业至少要投入不菲,即使临床顺利完成,按照230号公告,药品上市后3年内仍然要完成一致性评价,否则将注销药品批准文号。”
专家还表示,尽管理论上企业可以撤回已申报的老3类药物注册申请,按照新分类注册要求重新申报,但由于撤回申请代价较大,大多数企业可能会保持观望态度等待政策进一步明朗化。
“以往国内仿制药市场要拼速度、拼占坑的先来后到,现在逐渐有和美国仿制药市场接轨的趋势,即在仿制药质量、疗效与原研产品一致的基础上比拼价格,谁的价格低,市场占有率就高。”某药业经理认为,国内企业能否通过一致性评价将决定其在招标以及价格上的优势。”
不过,一位从事药品政策研究的教授告诉记者,通过一致性评价的仿制药优惠政策并没有作为硬性规定,具体落实有待观察。未来仿制药市场的优胜劣汰仍需要一段时间检验,拥有新剂型、新工艺等形成一定技术壁垒的仿制药企业可能会脱颖而出。
对此,某生物医药产业技术研究院有限公司总经理同样认为仿制药企未来将面临来自BE试验与审评的双重压力。一方面企业有意愿做BE试验,但受临床实验数据自查影响,临床机构的积极性不高,不愿意承担,BE试验无地可做;另一方面,仿制药仅排队待审评就长达2-3年,补充申请可能会进一步加剧注册积压的情况,再次导致审评时效不可控。他指出,通过集中审评的手段解决注册积压问题治标不治本,“应当从政策上引导企业理性申报,以品种的临床价值为衡量指标,达到企业理性申报,政府规范有效审评,即合理加快审评进度,完善审评制度等,形成审评进出品种平衡,申报审批良性发展,营造良好的医药研发环境,各方共同努力为医药行业的发展做出贡献。”

分享到:




 相关新闻
相关新闻













