



2012年7月,在《美国食品药品管理局安全与创新法案》(FDASIA)获得国会批准之后,突破性疗法认定(BTD)开始生效。新规为用来治疗严重疾病或者危及生命的疾病的药物提供了新的资格认定,制药公司要获得这种资格,需要通过初步临床证据表明,其研发的新药显示出了比现有药物有明显改善的治疗效果。
在FDA出台的一系列加快药物开发和批准计划中,BTD最初看似是一个相当不起眼的补充,外界对其所产生影响的预期并不大,在有些人看来,制药行业每年提出的突破性疗法认定申请也许少到只有两三件。然而,根据FDA发布的2013年行业指导报告,在这项立法生效之后的短短一年时间里,这一数字上涨了十倍。如今,在BTD计划进入的第三个年头,实际数字比最初的估计高了30~40倍。
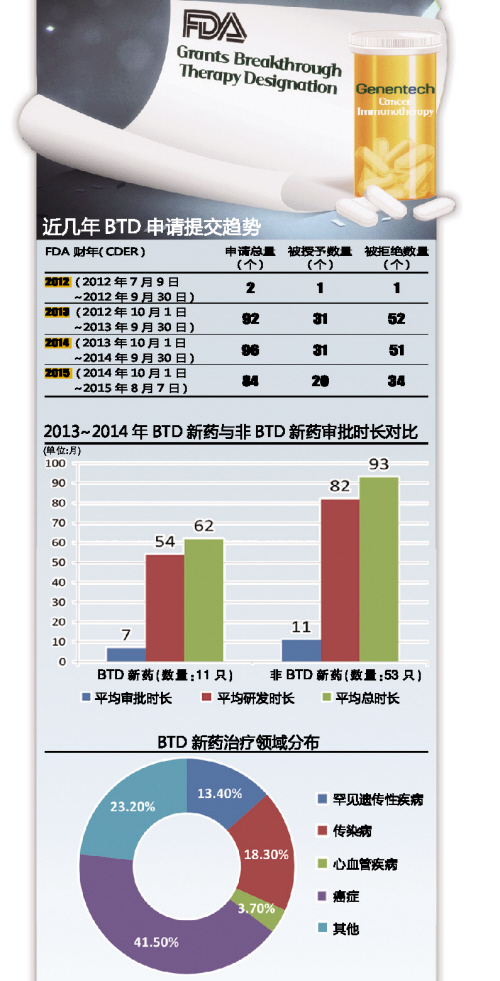
Q1:BTD审批提速是否实至名归?
从Ⅰ期临床试验阶段开始,BTD就为高效的药物开发提供了有力后盾,FDA各部门在跨学科的协同参与中各司其职
初看起来,BTD似乎就是FDA快速审批通道(Fast Track)的另外一个翻版。快速审批通道是FDA建立的一个独立的药品审评资格计划。不过,FDA很快发现,BTD所带来的好处要比快速审批通道更加广泛。从Ⅰ期临床试验阶段开始,BTD就为高效的药物开发提供了强有力的指导,尤其是其识别出了临床试验设计取得的进步(比如适应性临床试验),以及不断发展之中的技术(比如伴随式诊断)。
最值得注意的是,BTD向临床试验发起者提供了FDA的承诺,这一点被视为FDA在跨学科的协同参与中将会做到各司其职,这种参与不仅仅是在FDA部门层面,而且还跨越了各级管理层。
然而,FDA和临床试验发起者很快就遭遇到了挑战。就FDA方面来说,面临申请太多、时间太短的问题。目前,FDA解决的办法就是承诺给予一定的灵活性。FDA审评人员同意,制药公司在提交申请时,可以提供较少的稳定性数据,他们在审评周期内也可以接受研究数据的修改,并增加产品上市销售之后的相关承诺。FDA表示,其愿意在这个审评程序中进一步拓展这种便利性。
在遭到拒绝的BTD申请中,大约72%与试验设计或分析问题有关。这表明,试验发起者在提交申请之前,必须在与FDA的沟通交流上下功夫。与FDA相关评审部门进行非正式的磋商,可能会有助于试验发起者更好、更早地了解FDA到底需要什么样的数据。
在美国智库“布鲁金斯学会”最近举行的会议上,有人指出,这种早期的沟通交流既可以增强BTD申请的可行性,又可以减少那些毫无意义的申请数量。
对于临床试验发起者而言,挑战则在于其自身资源方面。根据BTD规则,产品的上市销售申请在Ⅱ期试验阶段之后被提交,这比传统的新药上市许可申请(NDA)提早两年,这种情况会因为制药公司在全球多个市场同时申请而进一步复杂化(包括国外监管机构对生产现场进行检查)。对生物制剂的申请来说,BTD可能还涉及到制药公司要以更加广泛的标准规范提交申请,这些标准规范在项目后期阶段(从临床试验基地的推出,以及转移到商业生产基地的上市销售之后)会变得更加严格。因此,制药公司必须在申请中包括产品获批之后的生命周期管理计划。
这可能引发的后果就是,如果制药公司过于频繁地对潜在的突破性药物投下赌注,这就会让自身的资源处于过度紧张状态。FDA也担心这一点,因为BTD计划广受欢迎,但立法并没有为满足更多的申请需求提供足够的资金支持。
Q2:BTD是否值得药企尝试?
2013~2014年, BTD新药的平均总开发时间比非BTD新药少2.5年
如果BTD带来回报,临床试验发起者会得到什么?平均来说,2013~2014年,新分子实体(NME)药物批准的BTD产品(数量为11只)的总开发时间要比非BTD新分子实体(数量为53只)少2.5年。换句话说,BTD本身带来了近3.5年的审批时间优势。
就BTD方面来说,这种初期的表现并没有被投资界所忽视。2014年,投入生命科学公司的资金占风险资本投资总额的18%,光是风险资本家就对私营生物技术公司投入了60亿美元。美国国会进行相关话题讨论时称,这种面向生物技术和制药领域的私营投资趋势是积极的监管和决策环境所带来的结果。
与此同时,广大患者肯定会对BTD计划取得的成果感到高兴:过去三年来,医药行业提交了300多件资格认定申请,有超过25只产品获得批准,其中包括在FDA 药物评价与研究中心(CDER)和生物制品评价与研究中心(CBER)获得批准的NME和补充剂,这些产品横跨多个治疗领域。
Q3:BTD是否为公众健康带来更大利益?
在此框架下,FDA能够支持从早期临床试验阶段到上市销售后风险管理阶段的医药创新,但需注意资源管理
FDA指出,BTD的推动力主要源于药物研发领域出现的几个新趋势,比如分子靶向治疗药物的兴起,其往往与伴随式诊断工具结合在一起。在这些靶向治疗药物中,有一些比现有药物治疗效果更大,这种效果甚至在对人体所做的初步试验中就表现得很明显。如果在药品开发的早期阶段,就能观察到药物在治疗严重疾病时产生了很大的效果,那么,实施更长时间、覆盖传统的试验全程的临床开发计划看上去也就显得多余了。
与传统审批程序所需的时间相比,FDA在尽早向患者提供前景看好的药物方面也面临着挑战,尤其是在患者没有其它药物可供使用的情况下。BTD似乎是一条更加符合FDA自身使命的路径:一方面要保护公众的健康,另外一方面要促进医疗产品的开发。BTD要求制药公司明确,其所开发的药物在具有临床意义的终点上比目前可供使用的药物表现出实质性的优势,此举既提供了胜过现有药物的优势,又提供了合理预测临床治疗好处的证据。
因此,这将让FDA拥有一种手段,用来解决其所认为的监管悖论:一方面,FDA对药物的诸多不确定性感到不快,另一方面,其又愿意接受一只药物在获批之前存在的某些未知数,这两者之间存在着固有但却可以调和的矛盾。以前,FDA是在药物开发的关键节点进行定期干预,但是现在,FDA的干预方式转向了“全覆盖、所有层次、每时每刻”的促进框架。通过这种做法,FDA能够支持从早期临床试验阶段到上市销售后风险管理阶段的医药创新。
不过,要让这种情况顺利实现,有关方面必须在资源层面采取相应的措施:要么临床试验发起者必须减少申请数量,提交更少或者更高质量的申请,要么FDA通过扩大BTD计划和收取一定的费用,扩大其审批规模。
 相关新闻
相关新闻川沙总部
地址: 上海市浦东新区川大路585号
邮编: 201299
电话: +86 (21) 5859-1500(总机)
传真: +86 (21) 5859-6369
海外:
Email: marketing@medicilon.com
Tel: +1 (617) 888-9294(U.S.)
Tel: 0044 7790 816 954 (Europe)
Tel: +82 70-8269-5849 (Korea)
Tel: +81 80-4421-6898 (Japan)















