7月14日,美国食品与药品管理局(FDA)发布了药物和诊断共同开发需遵循准则的指南草案,旨在将该指南作为实用的参考框架,帮助开发者推进治疗产品和伴随诊断的开发,并帮助FDA工作人员审查这些产品。
FDA在指南草案中写到,“IVD伴随诊断和治疗产品的共同开发对于精准医疗的推进至关重要。FDA试图通过向开发者提供一套准则,帮助他们进行有效的共同开发以及执行FDA的监管要求,进而加速精准医疗方面的创新。”
Rx/Dx共同开发方案的监管
对FDA来说,理想的Rx/Dx共同开发方案是,在药物研发的最早期确定需要开发的伴随诊断,两者同时开发并同时进入市场。
两年前FDA发布伴随诊断的指南时阐述了上述理想方案。但后来FDA意识到,对药物和伴随诊断同时进行评估并不总是可行,因此允许将伴随诊断的评估时间推后,先确定药物是否对某种无药可用的威胁生命的疾病有效。
通常情况下,FDA认为伴随诊断属于ClassIII、高风险产品,需要售前批准。但在共同开发指南中,FDA认为,一些Rx/Dx共同开发产品中的伴随诊断可归类为ClassII、中度风险产品,得到510(k)许可或de novo request后即可进入市场。
药物和检测开发的推进过程是明显不同的,该指南提供了一个图表,告诉开发者如何调整计划以及何时应该向FDA寻求意见。FDA建议,开发者需对治疗和诊断的研发都有一定的了解,并且治疗和诊断研发双方都要出席与FDA药物和诊断部门的会议。
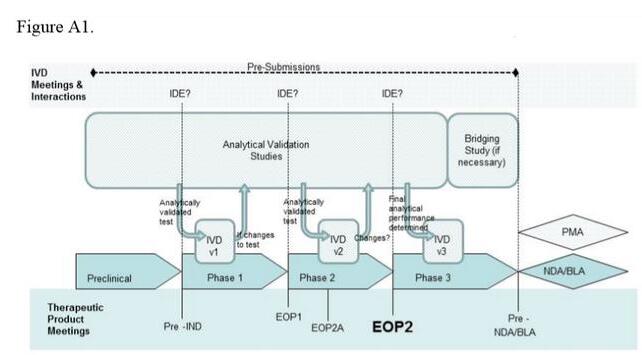
FDA对伴随诊断的定义是,一种必需的验证药物安全性和有效性的检测。因此,FDA建议,伴随诊断(CDx)的性能分析需在它应用在药物的临床试验前进行。当没有足够的数据证明一种试验性新药对患者的风险时,FDA会对这项研究下达“clinical hold(临床试验暂停)”通知。但FDA表示,伴随诊断性能分析的不确定性不会导致这种暂停。
如果一种药物的伴随诊断之前未得到FDA批准用于这种特定用途,开发者必须提出申请并获得器械临床研究豁免(IDE)。在该指南草案中,FDA概述了IDE申请应该包含的信息类型,以及在什么情况下体外诊断(IVD)可以不需要这个过程。该指南还讨论了检测开发者在利用训练样本集、检测设计变更的影响和IVD桥接试验中应该考虑的一些因素。
样本采集
近一段时间,FDA一直在告诫开发者,医生越来越多地预筛选病人,以明确他们进行生物标志物临床试验的资格,而这种行为会给Rx/Dx共同开发带来问题。FDA表示,“预筛查会产生具有偏好性的临床试验人群,这个群体不能代表真实世界中使用IVD伴随诊断的群体。因此,FDA强烈地反对挑选测试对象。”
FDA建议,开发者应该要求临床试验参与单位提交的样本来自于所有潜在候选检测人群,而不是经过本地检测筛选出来的人群。FDA说,这样才能够评估IVD真实的分析能力,并且能够确保意向治疗人群不具有任何偏好性。
该指南草案还表示,FDA将在很多方面更加灵活,例如,允许在早期研究中使用临床试验分析,可展示NGS检测中有代表性的标记物的分析验证结果,当真实的病人样本无法获取时可利用人为样本研究某种特定标记物。
但是FDA在指南中还指出,样本采集对一个共同开发方案的成功至关重要,鼓励开发者从所有招募的检测对象中获取样本。这样可确保临床试验分析进行不下去的情况下,开发者仍然能够对伴随检测进行验证且使其商业化。
生物标记物相关试验的设计
该指南指出,制药公司可以进行不同类型的生物标记物试验设计,一种设计是按照阳性和阴性生物标记物状态将患者随机分为两组,另一种设计是仅将阳性生物标记物状态的患者随机分到治疗组。
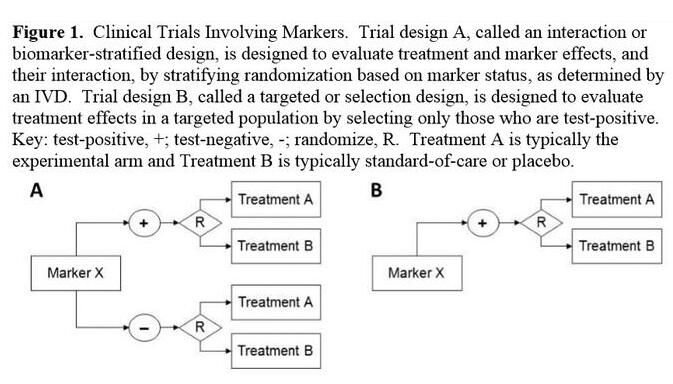
FDA表示,在衡量生物标记物的预后和预测价值方面,第一种试验设计最有用。但是当有证据表明生物标记物阴性的患者对治疗没有应答时,FDA会加速对只招募阳性生物标记物患者的精准药物研究的审批工作。FDA还讨论了一些方案,使制药公司在一项前瞻性研究完成后,可以基于生物标记物回顾性地评估病人的应答情况。
根据伴随诊断是如何在药物试验中使用的,它将获得一份声明文件表明它的作用,是用于预测疗效,监测药物剂量或者停药,还是用于筛选进行临床试验的病人。FDA表示,“就筛选病人的伴随诊断声明来说,如果主要药效试验表明,该药品对IVD筛选的人群具有足够的安全性和有效性,这就说明该IVD得到了临床验证,其选择的群体能从该治疗产品中获益。”
指南草案的意见征集
1998年,FDA批准了第一个乳腺癌治疗药物Herceptin(trastuzumab)和其伴随诊断HercepTest。近年来,Rx/Dx共同开发产品快速增多,尤其是肿瘤类产品。该指南草案中关于共同开发的建议都来自FDA多年来批准这类产品的经验。
个性化医学联盟(Personalized Medicine Coalition,PMC)的执行副总裁Amy Miller说,“FDA药品和诊断部门开发了内部流程,使肿瘤药物通过FDA审查。我们建议FDA利用这些经验,为开发者和FDA其他审查领域的员工起草一份“如何做”的指南,所以他们就制定了这份指南草案。”
该共同开发指南草案已经制定了很长一段时间了,多年来FDA与利益相关人士在研讨会和行业会议上就文件中的相关准则进行了交流。Miller说,PMC会进一步研究这份草案并提出改进意见。公众有90天时间对该指南草案提出意见。

























 相关新闻
相关新闻

 关于我们
关于我们