Emai:marketing@medicilon.com.cn
业务咨询专线:400-780-8018
Tel: +1(626)986-9880(U.S. - West Coast)
0044 7790 816 954 (Europe)
Email: marketing@medicilon.com
地址:上海市浦东新区川大路585号
邮编:201299
电话:+86 (21) 5859-1500(总机)
传真:+86 (21) 5859-6369





© 2023 上海美迪西生物医药股份有限公司 保留所有权利 沪ICP备10216606号-3











业务咨询
中国:
Email: marketing@medicilon.com.cn
业务咨询专线:400-780-8018
(仅限服务咨询,其他事宜请拨打川沙总部电话)
川沙总部电话: +86 (21) 5859-1500
海外:
+1(626)986-9880(U.S. - West Coast)
0044 7790 816 954 (Europe)
Email:marketing@medicilon.com




药物研发的主要思路是通过药物来抑制或激发疾病发病机制中的某一个靶点。但是,近年来系统生物学为不断发展的药物研发提供了一种新的思路:多靶点药物治疗。同时,对疾病发病机制的进一步研究和认识也表明了,单独靶点的抑制或激发,在复杂疾病治疗中有局限性。多靶点药物治疗,可使药物同时作用于多个靶点,对各靶点的作用可以产生协同效应,使总效应大于各单效应之和,达到最佳的治疗效果。
多靶点药物的概括
多靶点药物按组分的不同可以分为三种形式。其一为:多种药物联合用药,缺点是所含药物彼此间容易发生相互作用而产生不良反应。其二为:多组分单药片,即在一个给药单位中含有多种活性组分。其三为:某一单组分药物可以同时选择性作用于多个分子靶点,即严格意义上的多靶点药物。单组分药物在药物代谢上是较联合用药和多组分的药物有更多优势的,因其可以克服各组分相互作用产生的不良反应。
由于单靶点药物只能调控疾病过程中的一个点,而各种临床重大疾病通常是多重因素导致的结果,且其病理机制及疾病治疗过程非常复杂。因此,单靶点药物往往会有治疗效果不佳、药物筛选效率不高等缺点。而多靶点药物可以同时作用于多个病理环节、多种发病机制而产生协同作用效果,从而提高药物的疗效。
药物靶标通常处于多个信号通路中,具有多重生物学功能,过分激活或抑制某一生物靶标,在干预一种生物学功能的同时,也可以影响其它正常生物学功能,从而导致毒副作用的发作。多靶点药物可以更好的平衡多个病理因素间的关系,可以在相对更低的血药浓度水平,产生单靶点药物需要高浓度才能产生的生物学效应,且对生物靶标一般具有弱亲和力的特点,因而可以减少药物的不良反应。
生物机体是一个复杂的可自我调节和平衡的系统,长期使用某一单靶点药物治疗疾病,可以诱导机体内部的适应性变化从而激活对抗保护机制或者旁路代偿机制等,使疾病对该种药物不再敏感,造成药物的耐药性。而多靶点药物可以通过同时干预疾病的主要致病靶标及其代偿信号通路或者其保护性信号通路而减少疾病对药物产生的耐药性。
多靶点药物需要协调平衡多方面的参数使之处于适度区间,如药效学方面需要考虑靶标组合的合理性、活性的平衡性和靶标的选择性;药动学方面,要考虑ADME各方面特性的适当与否;在化学方面,要考虑其理化性质的合适与否等;靶标组合方面,它要求选择疾病病理机制中最关键的几个靶标进行组合,并保证靶标组合的合理性;活性平衡方面,多靶点药物对各靶点的作用强度接近而不宜差别太大;靶标选择方面,多靶点药物只选择作用于所确定的靶标组合,而不应对其他靶标有多余的活性,以减小不必要的副作用。
尽管存在很多问题和困难,随着现代系统生物学、化学生物学以及计算机辅助药物设计技术等的发展,多靶点药物的研究也在一步步的拓展。
已上市多靶点药物的概况
随着多靶点药物发现技术的不断成熟,已经有越来越多的多靶点药物进入临床应用,尤其是在癌症、糖尿病、病毒和细菌感染等复杂性疾病中。近年来,FDA先后批准了多个多靶点酪氨酸激酶抑制剂上市,包括2005年获批的索拉非尼、2006年获批的达沙替尼、2007年获批的舒尼替尼和拉帕替尼。另外,多靶点药物还包括心血管药物奥马曲拉、特波格雷、普齐地洛;中枢神经系统药物拉多替吉、奥氮平、卡巴拉汀等等。
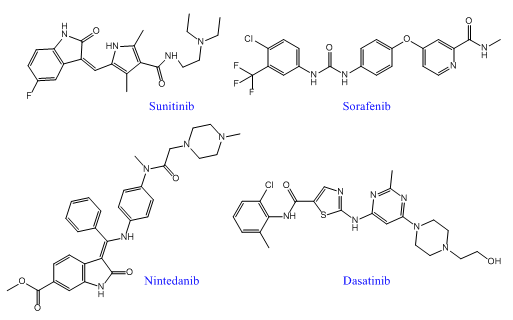
索拉菲尼是第一个用于肿瘤治疗的多靶点激酶抑制剂,能抑制RAF-1、B-RAF的丝氨酸/苏氨酸激酶活性,同时可以抑制VEGFR-2、VEGFR-3、PDGF-β、KTI、FLT-3等多种受体的酪氨酸激酶活性。索拉菲尼具有双重抗肿瘤作用,既可以阻断有由RAF/MEK/ERK介导的细胞信号转导通路,直接抑制肿瘤细胞的增殖,还可以通过作用于VEGF、PDGF-β等受体,抑制新生血管的形成和阻断肿瘤细胞的营养供应和代谢而达到遏制肿瘤生长的目的。在2005年12月,美国FDA就批准其用于治疗晚期肾癌,后2007年11月,美国FDA再次批准其用于无法切除治疗的晚期肝癌。该药在我国分别于2006年和2008年被批准用于晚期肾癌和晚期肝癌的治疗。2011~2015连续五年,索拉非尼的年销售额超过10亿美元。
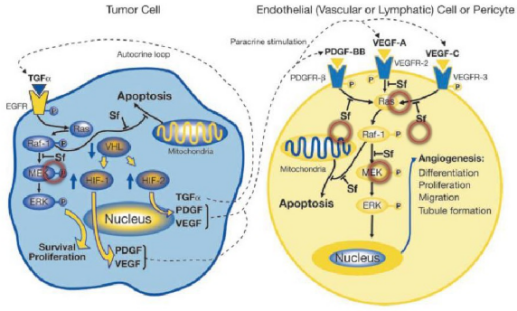
索拉非尼抗靶向细胞增殖和血管生成示意图
舒尼替尼是一种新型吲哚酮类口服、选择性多靶点酪氨酸激酶抑制剂,除抑制VEGFR-1、VEGFR-2、VEGFR-3、PDGFR-α、PDGFR-β的活性外,同时也抑制几种其他相关的酪氨酸激酶的活性,包括c-KIT、FLT-3和RET受体,具有抗血管生成和抗肿瘤活性的双重作用。美国FDA已经批准其用于晚期肾细胞癌,胃肠间质瘤和晚期胰腺神经内分泌瘤。临床前期研究表明,舒尼替尼能够有效抑制人NSCLC异种移植瘤模型的生长。多项临床研究评估了舒尼替尼在晚期NSCLC治疗中的作用,初步显示其在多线治疗后的晚期NSCLC中仍能取得一定的疗效,能改善患者生存,且毒性耐受。
阿西替尼是新一代强效的多靶点药物,其主要的作用靶点为VEGFR-1、VEGFR-2、VEGFR-3、PDGFR-β和c-KIT,是目前对VEGFR信号通路抑制率最强的酪氨酸激酶抑制剂,在多种实体肿瘤中显示出良好的抗肿瘤活性。FDA已批准用于晚期肾细胞癌的二线治疗。在一项阿西替尼单药治疗晚期NSCLC的II期临床研究中,显示了其在NSCLC患者中良好的抗肿瘤活性和安全性。
尼达尼布为三重酪氨酸激酶抑制剂,靶点包括VEGF、PDGF和FGF,也可抑制MAPK和AKT的激活。体外研究表明,尼达尼布能持续抑制VEGFR-2达30小时以上。I-II期临床研究显示,尼达尼布对第一、二线治疗失败的复发转移性NSCLC及局部晚期NSCLC有效,部分患者表现为肿瘤缩小,病情稳定。
新多靶点抑制剂的发现
抑制p53-MDM2蛋白结合和组蛋白去乙酰化酶(HDACs)是抗肿瘤药物开发的重要靶点。受MDM2和HDACs协同作用的启发,Shipeng He等人发现了第一个MDM2/HDACs双抑制剂(14d),对MDM2/HDACs这两个靶点都有很好的活性,且其抗肿瘤机制在癌细胞中得到验证,为新型抗肿瘤药物的开发提供了一种很有前途的小分子抑制剂[1]。
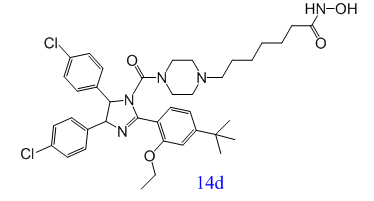
p53是一种转录因子,在预防肿瘤发展中扮演着重要角色。大约50%的人类癌症与p53的失活有关[2]。MDM2基因上的rs2279744位点发生突变后,其蛋白产物能够与 P53 蛋白结合并增强其降解,从而导致 P53 蛋白的抑癌作用减弱。因此,抑制p53-MDM2蛋白质结合成为了一种新兴有前途的癌症治疗策略[3]。研究发现,单一化合物同时调制多个靶点可能会产生更加卓越的功效以及更少的副作用[4]。组蛋白去乙酰化酶(HDACs),一种表观遗传酶,在调节肿瘤抑制基因的表达方面发挥着至关重要的作用[5]。然而,大多数HDAC抑制剂需要与其他抗肿瘤药物结合使用,实现协同效应[6]。因此,,Shipeng He等人合理研究设计了首个MDM2/HDAC双抑制剂(14d)。在A549异种移植模型中,双抑制剂14d的抗肿瘤效果良好,证明了这种新型多靶向抗肿瘤药物设计策略的价值。
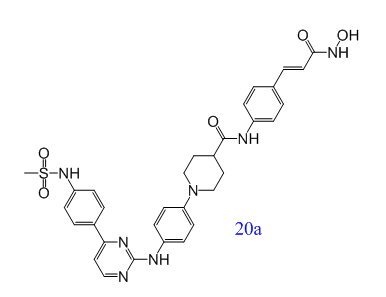
在临床上,白血病患者常因化疗疗效有限,且易受侵袭性真菌病原感染。Yahui Huang等人提出了一种新的治疗策略:小分子抑制剂可以同时治疗白血病和侵袭性真菌感染(IFIs)。新型Janus激酶2(JAK2)和组蛋白去乙酰化酶(HDAC)双重抑制剂对血液细胞系具有较强的抗增殖活性。其中,化合物20a是一种高度活性和选择性的JAK2/ HDAC6双重抑制剂,在几种急性髓系白血病(AML)模型中显示出极好的体内抗肿瘤作用,并可与氟康唑协同治疗耐药的白色念珠菌感染[7]。
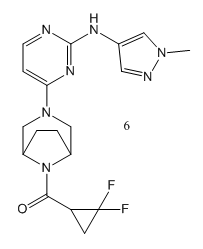
Christel J Menet提到小分子抑制剂6,它具有较好的选择性和良好的口服生物利用度,并在JAK1和TYK2的双重抑制给药方案中取得了临床益处。在最近的一篇论文中[8],研究结果表明,TYK2 / JAK1抑制剂6有在健康受试者和斑块性银屑病患者中有良好的安全性和耐受性,此结论支持抑制剂6在银屑病及其他炎性疾病治疗中值得进一步临床研究。
从目前药物研发的情势来看,创新药物的研发是迫在眉睫的,而提高药物研发的综合水平是我国所面临的主要难题。因此,在对已知靶点合理的、最大可能的利用是可实行且回报率高的研究方向,也应多关注于多靶点药物的设计,从而获得更多新的多靶点抑制剂,给疾病患者带来更好的治疗效果。
参考文献:
1,ShipengHe, Guoqiang Dong, Shanchao Wu, Kun Fang, Zhenyuan Miao, Wei Wang and ChunquanSheng.Small Molecules Simultaneously Inhibitingp53-Murine Double Minute 2 (MDM2) Interaction and Histone Deacetylases (HDACs):Discovery of Novel Multitargeting Antitumor Agents.J. Med. Chem. 61, 16, 7245-7260.
2,Hainaut,P. Hollstein, M. P53 and human cancer: the first ten thousand mutations. Adv.Cancer Res. 1999, 77, 81– 137.
3,Wang,S. Zhao, Y. Bernard, D. Aguilar, A. Kumar, S. Targeting the MDM2-p53protein-protein interaction for new cancer therapeutics. Top. Med. Chem. 2012,8, 57– 79.
4,Singh,A. K. Chauhan, S. S. Singh, S. K. Verma, V. V. Singh, A. Arya, R. K.Maheshwari, S. Akhtar, M. S. Sarkar, J. Rangnekar, V. M. Chauhan, P. M. S.Datta, D. Dual targeting of MDM2 with a novel small-molecule inhibitorovercomes TRAIL resistance in cancer. Carcinogenesis 2016, 37, 1027– 1040.
5,Ropero,S. Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol.Oncol. 2007, 1, 19– 25.
6,Ong,P. S. Wang, X. Q. Lin, H. S. Chan, S. Y. Ho, P. C. Synergistic effects ofsuberoylanilide hydroxamic acid combined with cispiatin causing cell cyclearrest independent apoptosis in platinum-resistant ovarian cancer cells. Int.J. Oncol. 2012, 40, 1705– 1713.
7,YahuiHuang, Guoqiang Dong, Huanqiu Li, Na Liu, Wannian Zhang and Chunquan Sheng.Discovery of Janus Kinase 2 (JAK2) andHistone Deacetylase (HDAC) Dual Inhibitors as a Novel Strategy for theCombinational Treatment of Leukemia and Invasive Fungal Infections.J. Med. Chem. 2018, 61, 6056−6074.
8,Banfield,C. Scaramozza, M. Zhang, W. Kieras, E. Page, K. M. Fensome, A. Vincent, M.Dowty, M. E. Goteti, K. Winkle, P. J. Peeva, E. The Safety, Tolerability,Pharmacokinetics, and Pharmacodynamics of a TYK2/JAK1 Inhibitor (PF-06700841)in Healthy Subjects and Patients With Plaque Psoriasis. J. Clin. Pharmacol.2018, 58 (4), 434–447.
 相关新闻
相关新闻川沙总部
地址: 上海市浦东新区川大路585号
邮编: 201299
电话: +86 (21) 5859-1500(总机)
传真: +86 (21) 5859-6369
海外:
Email: marketing@medicilon.com
Tel: +1(626)986-9880(U.S. - West Coast)
Tel: 0044 7790 816 954 (Europe)
Tel: +82 70-8269-5849 (Korea)
Tel: +81 80-4421-6898 (Japan)


 关于我们
关于我们