在美国,药价正成为头条新闻,并引发国会听证会的召开。在此背景下,生物类似药继续成为解决药价问题的希望。不过,希望并不等于现实。
人们对生物类似药寄予厚望,希望这类药物能够创造竞争,抑制不断攀升的生物药价格。但是目前这类药物仅占美国药费支出的20%左右,这与人们对其的希望不相符合。所以,生物类似药首先必须设法抢占市场。
迄今为止,FDA共批准了2个生物类似药:山德士(Sandoz)的Zarxio,是优保津(Neupogen,非格司亭,安进)的生物类似药;Celltrion的类克(Remicade,英夫利西单抗,JanssenBiotech)生物类似药Inflectra即将通过辉瑞在美国销售。(见2015年3月9日和2016年4月6日的BioWorldToday)
目前美国市场上正在销售的生物类似药只有Zarxio,该药于2015年9月上市。根据《生物制品价格竞争和创新法案》(BiologicPriceCompetitionandInnovationAct,BPCIA)的规定,生物类似药的商业化上市必须有180天的通知期,所以哪怕Celltrion和辉瑞成功地挑战了Jassen的类克仅存的一项专利,认定其无效,Inflectra也至少需要等到2016年10月2日才有可能上市销售。
类克仅存的这项名为471的专利将于2018年下半年到期,目前美国专利和商标局正在对其进行重新审查。之前挑战这个专利的所有企图均被驳回。Jassen是强生旗下公司,该公司也在美国联邦法院起诉Celltrion的生物类似药对其造成了专利侵权,并且一而再地表示,它将有力地捍卫自己的专利。美国麻萨诸塞州地方法院计划在2017年2月审理这个案件,但是鉴于Inflectra即将上市,Jassen已经请求将审理提前至2016年9月。
2个获批产品,1个上市产品,美国生物类似药市场远远落后于欧洲。早在十年前,欧洲药品管理局(EMA)就通过了全球首个生物类似药审批路径。之后其他一些国家的监管机构就沿用了EMA的监管方法,而不是自己建立一套独特的生物类似药审批路径。
2010年,FDA获得国会批准可以创建一个生物类似药的专门审批路径,但是因为BPCIA,FDA不能简单地模仿EMA,必须自己建立的一套专门的路径。因此,FDA不得不考虑,如何在同时满足简化和科学的要求下,在复杂的生物药审批途径之外,设立一个新的监管路径。
获得的数据
FDA一直在思考,产业界一直在行动。FDA药品审评和研究中心(CDER)JanetWoodcock最近对众议院说,这意味着FDA可能马上就将面对生物类似药的爆发。(见2016年2月5日BioWorldToday)
截至2015年9月30日,FDA已经收到了22个按照生物类似药简化审批途径(351(k))提交的新药临床研究(IND)申请。除了已经批准的2个药物以外,至少有6个生物类似药的351(k)IND申请已经被FDA接受。未来,一旦可互换开发路径出台,FDA有望每年接受5个新的351(k)申请和2个可互换申请。(见2015年7月1日BioWorldToday)
以上预测可能还是保守估计。FDA的一名发言人告诉BioWorldToday,总体上看,截至2016年4月18日,FDA有60个生物类似药项目在等待审批,已经发出的相关会议请求涉及19个不同的生物药参比品。BioWorld的报告称,还有更多的候选药物正处在早期的发现阶段。
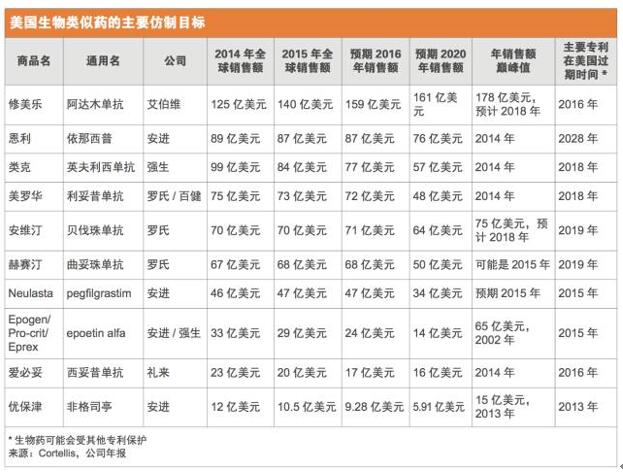
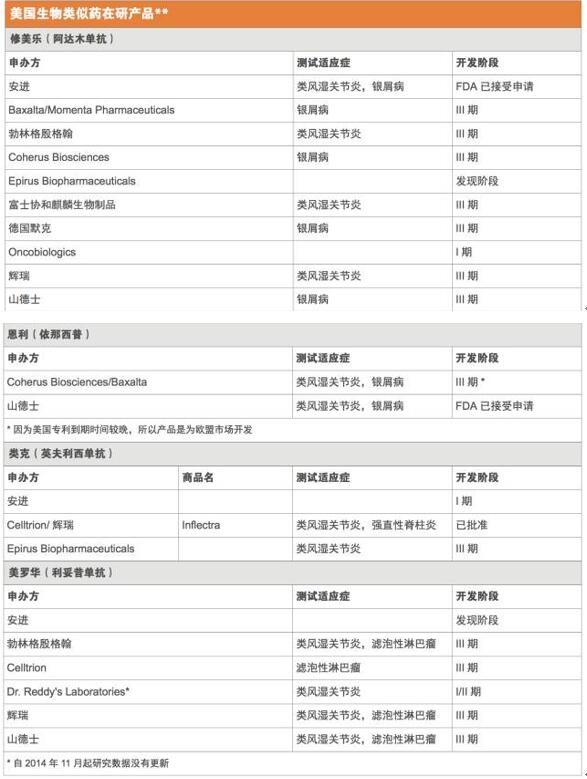
正如预期的那样,在研的生物类似药瞄准的都是最畅销的那些生物药,而且这些原研药中至少有一半大多数专利将在未来几年内到期。2015年全球最畅销药物艾伯维(AbbVie)的修美乐(Humira,阿达木单抗),当年销售额高达140亿美元,现在正理由当然地成为了仿制的目标。FDA已经收到了1个以修美乐为参比品的351(k)申请,另外,还有至少7个其他公司正在美国开展阿达木单抗生物类似药的III期试验。(见美国生物类似药的主要仿制目标和美国生物类似药在研产品)
除非FDA加快速度,否则该机构将面临生物类似药申请的积压。虽然FDA提前了将近2个月批准了Zarxio,但是审批Celltrion的生物类似药花费的时间要长得多。Celltrion于2014年8月8日提交了351(k),原本计划能在一年内完成审批。与这个时间表相配合,FDA安排在2015年3月召开专家委员会会议对该申请进行评审,但后来这个会议被取消,要求韩国公司Celltrion提交更多的数据。最后专家委员会会议直到2016年初才得以召开。
Celltrion并不是唯一一个遭遇监管延期的生物类似药申请者。FDA接受的351(k)申请中,有3个(参比品均为安进公司产品)的审评时间都超过了预定的10个月。

延迟的背后
延迟的原因,部分是因为生物药的复杂性。另外,FDA承诺将为这一新类型的药物建立起一个阶段式的、基于证据的监管路径也是原因之一。ProskauerRoseLLP生命科学专利事务的董事长SiegmundGutman告诉BioWorldToday,“他们正在处理的是真正艰难的问题,他们希望能够确保这些问题得以正确的解决。”
Gutman说,因为每个候选药物都代表着不同的技术问题,FDA需要在个案的基础上,逐一处理申请并对开发项目提出咨询意见。他补充道,所以,FDA没有在规定的审评截止日期完成审评,并产生申请积压。
FDA长期以来的经费问题也是造成审批延迟的原因之一。国会通过了BPCIA,将其作为平价医疗法案(AffordableCareAct)的一个部分,但是国会并没有为建立专门针对生物类似药的监管路径提供任何资金。因此,FDA不得不将现有的资源拿出来用在扩大和培训人员上,以实施这项法律,拟定指导草案,审评新的351(k)申请,以及制定教育医生和公众生物类似药相关知识的材料。
几年前,美国国会授权FDA收取生物类似药开发使用者费,为建立生物类似药审评路径提供部分资金,试图解决经费问题。2013财年开始,FDA开始收取这笔费用,当年该机构共收到约600万美元生物类似药费用。现在这笔钱正在稳步增加,2015财年超过了2000万美元。
使用者费用正在为生物类似药审批提供急需的资金,但是FDA的生物类似药审批工作仍然缓步慢行。2015年FDA完成了4个生物类似药指南中的第一个。然而,可互换性等问题的指南还只是草案,而且还必须最终完成一些关键问题的指南,比如生物类似药的命名,为支持生物类似性而必须提供的临床药理数据。
虽然FDA仍旧继续向前努力,但是来自于产业界和付费方的压力正日益增加。产业界和付费方担心,监管延迟、法律问题和医保的问题会在这个市场还未充分形成前就带来寒意。
——BioWorldToday,2016年4月25日









 相关新闻
相关新闻










